


Pasteur Eye Hospital
Eye Conditions
Contact Us

Retinitis pigmentosa (RP) is a group of inherited eye diseases that can lead to significant vision loss. These diseases affect the retina, the tissue at the back of the eye, where photoreceptor cells called rods and cones gradually lose their function, leading to sight impairment over time.
RP typically begins in childhood, but the onset and progression of symptoms can vary widely. Most individuals with RP experience a significant loss of vision by early adulthood and are often legally blind by age 40.
Common symptoms include:

RP is caused by mutations in over 60 different genes. These genetic mutations can be inherited in three primary ways:
Diagnosing RP involves several specialised tests:
If RP is diagnosed in one family member, it is recommended that all family members undergo screening.
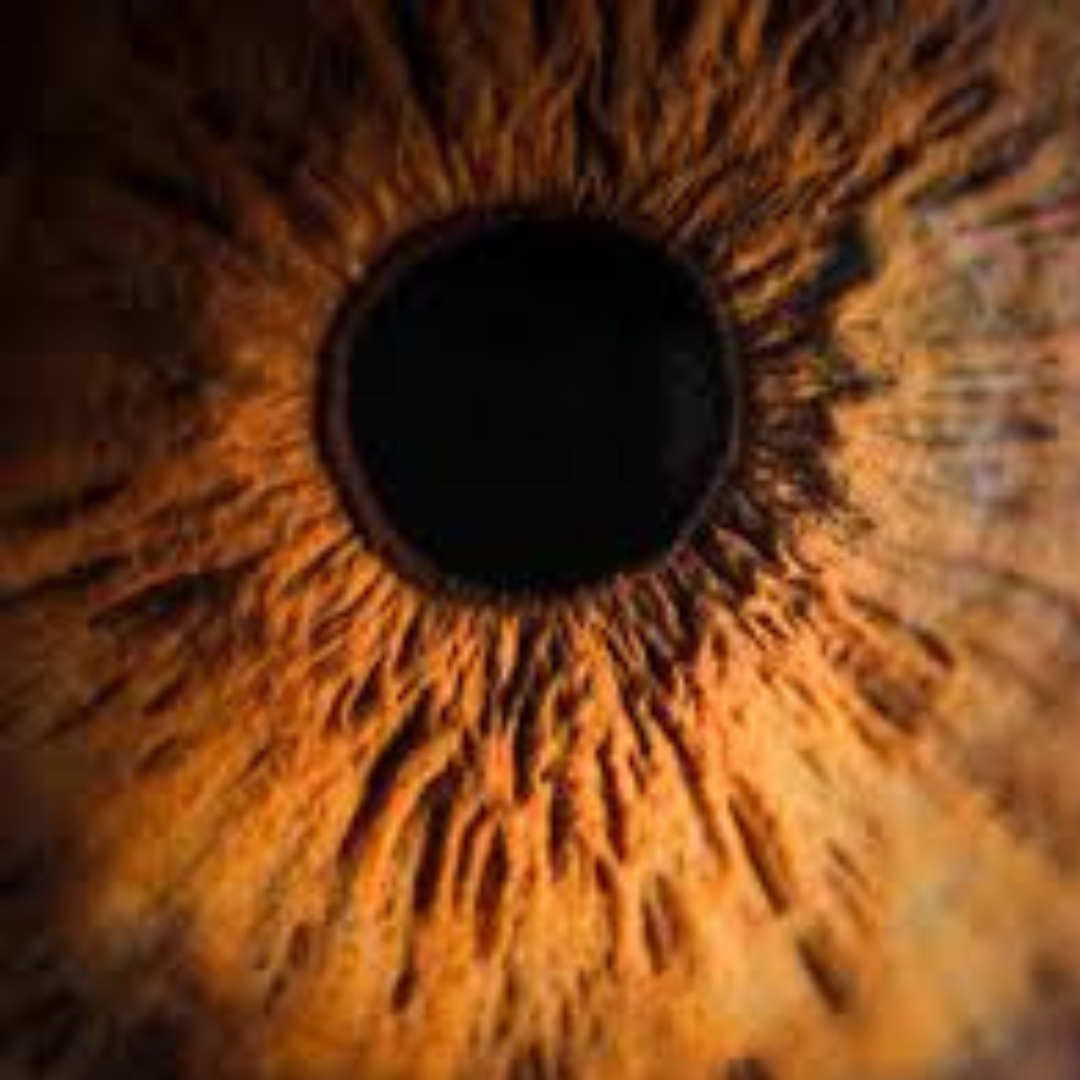
Retinitis pigmentosa is an inherited eye disease that causes progressive vision loss by damaging the retina’s photoreceptor cells.
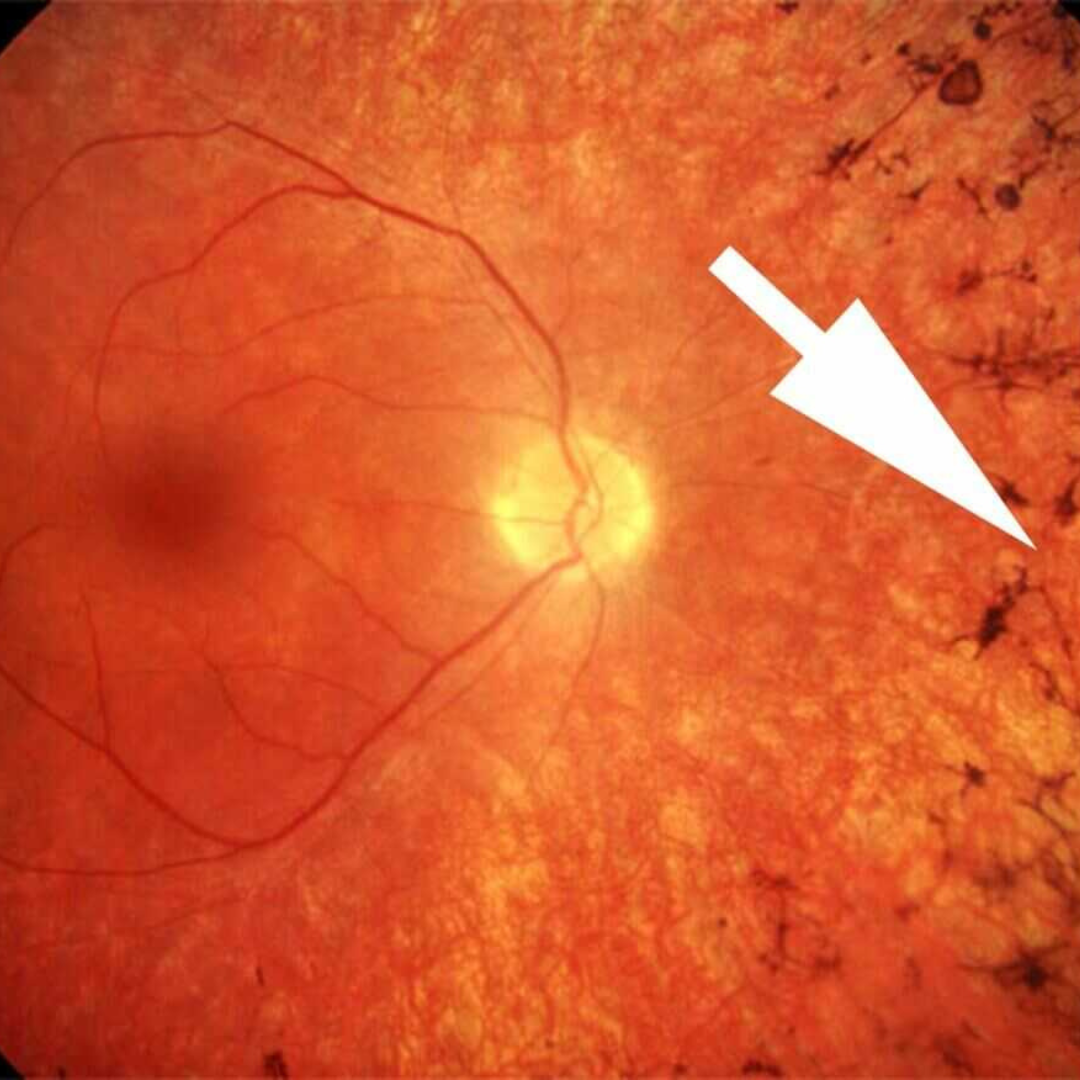
Retinitis pigmentosa (RP) causes gradual vision loss by damaging the retina’s photoreceptor cells, leading to eventual blindness.
While there is no cure for RP, some treatments can help manage symptoms:
Other emerging treatments include:
Please note that these treatments are still in experimental phases and not yet available.
Additionally, various devices and rehabilitation services can help individuals with RP maximize their remaining vision and maintain independence.
Retinitis pigmentosa is a serious condition requiring prompt and ongoing medical attention. If you or a family member has been diagnosed with RP, consult your eye doctor for personalized care and management strategies.
Pasteur Eye Hospital
Eye Conditions
Contact Us
© Copyright 2022 Pasteur Eye Hospital. All Rights Reserved.
No article or picture may be reproduced\published without the written consent of Pasteur Eye Hospital.
Managed with ❤️ by Cuberoo